Около 40 % мужчин и женщин в России имеют повышенное артериальное давление (АД). Среди хронических неинфекционных заболеваний артериальная гипертензия (АГ) — одно из самых распространенных [1, 2]. Артериальной гипертензией страдают около 35 млн человек, ежегодно выявляется до 0,5 млн больных, 30–40 % больных АГ не знают о своем заболевании [3, 4]. Повреждение органов-мишеней имеет важное значение в формировании гипертонической болезни (ГБ), занимает центральное место в его клинической картине, определяет диагноз, прогноз и исход.
Одной из наиболее значимых проблем в своевременной диагностике ГБ следует считать отсутствие субъективной манифестации. Повышенное АД является случайной находкой при диспансерном, профессиональном отборе, при определении показаний к санаторно-курортному лечению. M. Moser характеризовал ГБ как «безмолвный, тихий убийца» [5]. Надо считать закономерным, что ГБ начинается бессимптомно или мало- симптомно [6].
Несмотря на прогрессивное развитие экспериментальной и клинической базы знаний, имеют место затруднения в диагностике и интерпретации субъективных, объективных и инструментальных признаков сердечно-сосудистых заболеваний [7]. В этой связи можно задуматься о том, достаточно ли изучения только типичной картины заболевания и достаточно ли существующих традиционных объективных методов исследования и инструментальных подходов.
Особого внимания заслуживает анализ вероятных причин феномена малосимптомных форм течения ГБ, среди которых можно выделить следующие группы: 1) функциональные (достаточный уровень кровотока и лимфооттока для вымывания и предупреждения образования болевых субстанций; медленное развитие процесса; адекватная компенсация); 2) нейрогенные (повышение порога болевой чувствительности); 3) другие (восприятие болей зависит не только от выработки сигналов, но и от модуляции их во внутренних ганглиях органа, подкорковой и корковой обработки). Ниже мы представляем вероятные механизмы исходного формирования или последовательного развития субъективно неманифестированной формы гипертонической болезни.
Гипертензивно-ассоциированная гипалгезия
В ряде исследований установлено, что повышенное артериальное давление у человека и животных ассоциируется со снижением болевой чувствительности [8]. Возрастание толерантности к боли у больных ГБ впервые было показано в работе [9]. Эти исследователи оценили субъективное восприятие боли по значениям неинвазивной градуированной электрической стимуляции пульпы зуба (tooth-puip test). При обследовании 21 больного ГБ и 34 нормотензивных добровольцев установлено, что больные ГБ в среднем имели более высокий болевой порог. Эти значения были подтверждены другими методами: измерение порога полисинаптических компонентов мигательного рефлекса после поверхностной электрической тригеминальной стимуляции, измерение болевого порога после кожной электрической стимуляции и термальной стимуляции. Хотя механизмы, ответственные за гипертензивно-ассоциированную гипалгезию, остаются неизвестными, некоторые аспекты уже объяснены. Определенная роль отводится барорефлекторной системе, которая модулирует ноцицептивную передачу. Ослабление или прерывание синоаортальных афферентных импульсов различными методами заметно ослабляет или полностью устраняет гипертензивно-ассоциированную гипалгезию, что было доказано в различных экспериментальных моделях при остром или хроническом повышении давления [10, 11]. Кроме каротидноаортальных барорецепторов в процесс модуляции боли могут вовлекаться кардиопульмональные рецепторы, которые при увеличении объема циркулирующей крови (ОЦК) индуцируют рефлекторную гипалгезию, что может частично устраняться правосторонней ваготомией [12].
Исследования на людях и животных показали, что стимуляция отдельных участков головного мозга, первично серого вещества среднего мозга (центральное серое вещество вокруг водопровода), индуцирует глубокую аналгезию [13]. Полученные Randich и соавт. данные свидетельствуют о том, что в процесс антиноцицепции, индуцированной вагальной афферентной стимуляцией, вовлекаются структуры ствола мозга, служащие также для регуляции барорефлекторной активности и вазомоторного тонуса. Некоторые данные позволяют сделать заключение, что гигантоклеточные ядра ретикулярной формации (ядра Кахаля и Даршкевича) могут быть местом, специфически вовлеченным в процесс модуляции болевой чувствительности, связанной с изменением артериального давления, и ангиотензин III может играть роль медиатора в этой модуляции [11]. Другие факторы, вовлеченные в процесс антиноцицепции, вероятно, индуцированы острым повышением АД при введении катехоламинов [14].
Механизмы, регулирующие деятельность антиноцицептивной системы, делятся на нервные и гуморальные.
Нервные механизмы реализуются через ретикулярную формацию, таламус и серое вещество вокруг Сильвиева водопровода.
Гуморальные механизмы включают продукцию эндогенных опиатоподобных соеди- нений с выраженным обезболивающим эффектом — эндорфинов, энкефалинов.
В настоящее время на основе накопившихся экспериментальных данных можно говорить о наличии доказательств того, что существует, по крайней мере, 4 анальгезические системы.
Одним из важных механизмов подавления боли считается влияние нейронной опиатной анальгезической системы, основными структурными звеньями которой являются центральное серое вещество и ядра шва продолговатого мозга. Активация этих нейронов приводит к выделению нейропептидов (энкефалинов, эндорфинов) и моноаминов (серотонина, норадреналина), угнетающих передачу нервных импульсов от сенсорных нейронов на первые переключающие нейроны центральной нервной системы (ЦНС) в местах распределения нейронов, несущих болевую информацию (задние рога серого вещества спинного мозга, сенсорные ядра тройничного нерва) [15].
Другая опиатная анальгезическая система — гуморальная. Возникновение субъективно неманифестированной ГБ при этом может быть обусловлено эффектом эндогенных опиатов (эндорфинов, энкефалинов, динорфинов), биогенных аминов (серотонина, гистамина, катехоламинов) и γ-аминомасляной кислоты. Модулирующая активность всех этих субстанций, и особенно эндогенных опиатов, может быть одним из факторов, обусловливающих различную способность больных ГБ воспринимать боль [16, 17].
Функционирование гормональной неопиатной анальгезической системы сводится к тому, что в определенных физиологических и патологических ситуациях, сопровождающихся выраженным состоянием стресса, может возникать анальгезия, которую связывают с выбросом гормонов гипофиза. Об этом свидетельствует тот факт, что удаление гипофиза резко ослабляет анальгезию, связанную со стрессом. В эксперименте такую анальгезию вызывают введением животному инсулина, также стимулирующего гиперпродукцию гормонов гипофиза, прежде всего адренокортикотропный гормон (АКТГ), и вазопрессина. Кроме того, имеются сведения о выраженном антиноцицептивном действии соматостатина, нейротензина, однако пути их влияния и механизмы действия еще не совсем изучены [18, 19].
В настоящее время современной нейрофизиологией накоплен фактический материал, который свидетельствует о том, что многие агенты, выделяющиеся в ответ на стресс, в частности кортикотропин-релизинг фактор (КРФ), АКТГ, бета-эндорфин и кортикостероиды, оказывают антиноцицептивный эффект.
В литературе на основании анатомических, физиологических поведенческих исследований высказано закономерное предположение, что КРФ может играть глобальную роль в стрессорной реакции, участвуя не только как нейрогормон в тропных функциях гипофиза и инициируя АКТГ, но и как нейромодулятор и даже нейромедиатор в экстрагипофизарных циклах, способных в условиях стресса интегрировать все компоненты ответной реакции, изменяя нейрональную активность в ядрах мозга, которые вовлечены в вегетативных и поведенческих аспектах в стрессорные реакции, включая учащение сердечного ритма, повышение АД, повышение адреналина [20]. Большое значение в модуляции болевых стимулов имеют ядра raphe magnus, которые предположительно содержат болевые нейроны, отвечающие на «вредные» стимулы возбуждением активности (called ON cells), и другие, отвечающие понижением активности (called OFF cells). В серии наблюдений отмечено, что эти нейроны показывают спонтанные колебания нейроактивности, которые противо- положны (ON cells) и параллельны (OFF cells) спонтанным изменениям АД. Тот факт, что изменение нейральной активности предшествует изменениям АД и сохраняется после кардиопульмональной деафферентации, свидетельствует о том, что изменение нейральной активности необязательно требует барарецепторного афферентного импульса и эти оба процесса связаны с регуляцией АД. С другой стороны, изменения АД, индуцированные различными процедурами (инъекцией фенолефрина, нитропруссида или окклюзией абдоминальной аорты), изменяют активность ON и OFF cells через синоаортальные баро- рецепторы. Все вышеперечисленные данные свидетельствуют о близкой связи центров модуляции боли и кардиоваскулярной регуляции [13].
Обширные исследования посвящены эндогенным опиоидным системам. Результаты части из них показали, что гипертензивно-ассоциированная гипалгезия может быть уменьшена опиоидным антагонистом налоксоном. McCubbin и Bruehl наблюдали, что «предлечение» налоксоном заметно снижает силу связи между повышенными уровнями АД и уровнем болевой чувствительности у нормотензивных пациентов. Кроме того, найдены существенные различия в уровнях в-эндорфина плазмы у нормотензивных пациентов и больных ГБ и различия между нормотензивными пациентами с низкой и высокой толерантностью к боли [21, 22].
Таким образом, можно сделать вывод, что барорецепторная модуляция болевой чув- ствительности представляет особый аспект и заключается в более широких способностях барорефлекторной системы понижать активность процессов в ЦНС; этот механизм может работать, в дополнение к брадикардии и вазодилатации, в направлении предупреждения острого подъема артериального давления [23–25].
Наконец, следует сказать, что существование функциональной связи между АД и путями регуляции боли играет огромную роль в адаптивных механизмах организма к стрессу. Взаимодействие кардиовасклярной и соматосенсорной систем может быть представлено лишь как часть комплексного, координированного ответа организма на стрессовую ситуацию [26].
Причины этого феномена еще не до конца расшифрованы. В частности, неизвестно, является ли гипалгезия следствием гипертензии или в ее основе лежат общие, пока не идентифицированные механизмы [27–31]. В результате понижения АД необязательно происходят изменения в болевой чувствительности, что говорит о непростом взаимодействии этих двух факторов. Ghion S. и соавт. [29, 30] показали, что прием в течение 3 месяцев диуретиков и β-блокаторов и соблюдение низкосолевой диеты способствовали стабилизации цифр АД, но не привели к изменениям болевой чувствительности. С другой стороны, было показано, что лечение кетансерином индуцирует глубокую редукцию болевого порога [32].
Интересные данные получены Morley и др. [33], которые выявили повышение болевой толерантности при градуированной электрической стимуляции пальца больных ГБ и сопутствующим сахарным диабетом (СД) при сравнении с нормотензивными диабетиками.
Клинически важным следствием этого феномена, возможно, является бессимптомность или малосимптомность течения ГБ. Кроме того, доказано, что у лиц с повышенным АД по сравнению с нормотомиками на 30 % чаще отмечаются безболевые эпизоды ишемии на фоне повседневной активности и почти в 2 раза чаще — бессимптомное течение инфаркта миокарда [34].
По нашему мнению и по данным литературы, вероятные группы больных с малосимптомными проявлениями могут быть представлены следующим перечнем: 1) больные с сопутствующей ишемической болезнью сердца (ИБС); 2) больные после аортокоронарного шунтирования (АКШ), ангиопластики и стентирования коронарных артерий; 3) пациенты с сахарным диабетом; 4) пожилые люди; 5) больные, принимающие препараты, влияющие на нервную передачу; 6) больные энцефалопатией любой этиологии; 7) пациенты с психологическими особенностями личности (агностики).
Течение ГБ у больных с сопутствующей ИБС
В многочисленных эпидемиологических исследованиях приведены убедительные результаты, подтверждающие влияние ГБ на частоту развития атеросклероза, ишемической болезни сердца. По данным различных авторов доля больных ГБ с сопутствующей ИБС составляет 50–70 %. ГБ сопровождается прогрессирующим атеросклерозом венечных артерий сердца [35].
Одним из механизмов развития бессимптомных форм ГБ может быть развитие патохимических и патоморфологических изменений в миокарде, имеющее в своей основе атеросклеротическое стенозирование коронарных артерий. В результате этого происходит интенсивное раздражение окончаний афферентных нервов, которое наблюдается при внезапно развившемся нарушении коронарной перфузии. На уровне спинного мозга не успевает активироваться защитная система, предотвращающая раздражение восходящих путей, которые несут информацию в ЦНС [36]. Информация о повреждении тканей воспринимается нервной системой не пассивно. Уже на уровне первых синапсов сенсорных путей она регулируется с помощью сложных модуляторных систем.
В результате этого ЦНС запускает защитные механизмы, благодаря которым осуществляется серия рефлекторных реакций, направленных на прекращение дальнейшего действия болевого стимула. В условиях продолжающейся болевой реакции данная система обеспечивает приспособление всех основных систем и органов к деятельности организма в условиях длительной болевой стимуляции [15]. Указанные процессы могут способствовать понижению болевой импульсации при ассоцииро- ванных ГБ и ИБС.
Учитывая, что большая часть восходящих вагусных волокон находится в миокарде задней стенки левого желудочка, некоторые авторы считают, что возбуждение вагусных афферентных волокон может опосредованно влиять на ноцицепцию, повышая порог восприятия болевого ощущения [36].
Передача ноцицептивной импульсации из миокарда в ЦНС осуществляется по симпатическим нервам. Наряду с этим в эксперименте было показано, что усиление потока восходящих импульсов, идущих от сердца по блуждающему нерву, способно инициировать противоболевую систему ствола мозга, в частности, серотонинергические нейроны большого ядра шва. Активация этих нейронов увеличивает их тормозное влияние на спинно- мозговые вставочные нейроны, которые перерабатывают болевые стимулы [37].
При изучении порогов болевого ощущения у больных с ИБС и пациентов с болями в грудной клетке некоронарогенного генеза было выявлено, что величины болевого порога рефлекторного ответа у больных со стенокардией напряжения в 3 раза меньше, чем у здо- ровых лиц и у пациентов с кардиалгиями [38].
Прогностическая значимость безболевой ишемии миокарда (БИМ) при АГ была изучена при обследовании 407 больных в возрасте 40 лет. У больных с повышенным артериальным давлением и «нормальной» ЭКГ покоя, не предъявлявших жалоб и имевших положительные результаты нагрузочных проб на тредмил и сцинтиграфии, предсказа- тельная ценность БИМ в развитии инфаркта миокарда или внезапной смерти достигала 67 % [39].
При ИБС в нервном аппарате наступают значительные изменения в виде очагового выпадения и повсеместного снижения плотности холин- и адренергических нервных сплетений. Так, в околососудистых нервных сплетениях развиваются деструктивные явления как миелиновых, так и безмиелиновых волокон, при этом характерным является отсутствие в них катехоламинов и резкое снижение активности ацетилхолинэстеразы. Некоторыми исследователями показано, что процесс атеросклеротического поражения венечных артерий сопровождается явлениями деафферентации и десимпатизации как стенки артерии, так и миокарда в зоне ее кровоснабжения [40]. Вышеперечисленные изменения могут приводить к бессимптомному течению ассоциированных ИБС и ГБ. Особое значение при этом имеет факт предсуществующего течения субъективно неманифестированной ИБС или безболевой ишемии миокарда.
Важную роль в потере болевой чувствительности играют последствия операций на сердце. Так, через некоторое время после АКШ или транслюминальной баллонной коронарной ангиопластики (ТБКА) со стентированием признаки ишемии могут появиться вновь вследствие окклюзии шунта или стента, рестеноза или прогрессирования атеросклероза. Ангиографически выявленный рестеноз (часто определяемый как повторный стеноз 50 % и более) не всегда проявляется клинически, и частота его появления всегда выше, чем частота клинического рестеноза. Развитие клинических симптомов в сроки свыше 9 месяцев после ТБКА чаще бывает обусловлено прогрессированием атеросклероза коронарных артерий, нежели рестенозом. Иногда, особенно после хирургической реваскуляризации, рецидив ишемии может проявляться нетипичными признаками. Установлено, что повышенное количество случаев «немой», безболевой ишемии наблюдается именно у послеоперационных больных [41–43]. Пациенты после выполнения АКШ, у которых развилась послеоперационная стенокардия, представляют собой особую категорию больных, которым требуются тщательные оценка и лечение. В долгосрочном исследовании 977 больных после операции АКШ у 30 % наблюдалась стенокардия в течение первого года, у 46 % — по прошествии 3 лет и у 50 % — по прошествии 8 лет [44]. Более того, у многих послеоперационных больных наблюдается безболевая ишемия миокарда [45].
Механизмы сердечного болевого синдрома при ГБ многообразны. Они могут иметь среди прочих ангинозную природу, поскольку в условиях повышенного АД возрастают нагрузка на левый желудочек и его потребность в кислороде. Вышеперечисленные данные свидетельствуют о возможном влиянии АКШ и стентирования на течение не только ИБС, но и ГБ, с формированием феномена отсутствия субъективной манифестации повышенного АД.
Патогенетические механизмы развития субъективно неманифестированной формы ГБ у больных СД
В настоящее время модель развития диабетической нейропатии (ДН) представляет собой многостадийный процесс, включающий целый каскад патогенетических механизмов.
1. Полиоловый путь, снижение активности Na/K-АТФазы.
Персистирующая гипергликемия активизирует полиоловый путь утилизации глюкозы, что приводит к накоплению в нервной ткани сорбитола, фруктозы и снижению активности протеинкиназы С. Механизмы, посредством которых эти вещества могут повреждать нейроны и их отростки, до конца не ясны. Снижение содержания миоинозитола и активности Na+ /K+ -АТФазы приводит к ретенции Na+ , задержке жидкости, внутриклеточной гипергидратации миелиновой оболочки, снижению числа глиальных клеток аксонов и, в конце концов, к дегенерации периферических нервов и гипоалгезии, что может наблю- даться при сочетании ГБ с СД.
2. Гликирование белков основано на способности глюкозы, фруктозы и галактозы вступать в реакции гликозилирования с аминогруппами, входящими в структуру белков, липидов и нуклеиновых кислот. Гликозилирование различных молекулярных структур нейронов способствует аксональной атрофии, нарушению аксонального транспорта, демиелинизации нейронов и, как следствие, снижению проводимости, а значит, и повышению болевого порога, что и наблюдается у больных с сахарным диабетом и ассоциированной ГБ.
3. Нарушение обмена ненасыщенных жирных кислот, в первую очередь дигомо-γ- линоленовой и арахидоновой, ведет к нарушениям в циклооксигеназном цикле, снижению продукции вазоактивных субстанций и, в результате, к ослаблению эндоневрального кровотока. Это может способствовать понижению эффективности нервной передачи, в том числе и болевых стимулов.
4. Нарушение синтеза нейротрофных факторов и/или экспрессии их рецепторов.
5. Окислительный удар. Избыточное образование свободных радикалов с последующим повреждением мембранных структур нейронов и ДНК ведет к нарушению функций нервных клеток. Помимо прямого повреждающего действия накопление свободных радикалов способствует нарушению энергетического обмена, развитию эндоневральной гипоксии (рисунок).
Поражение афферентных висцеральных нервов, идущих от сердечной мышцы, приводит к тому, что ишемия/инфаркт миокарда могут протекать без боли. У больных СД каждый третий инфаркт миокарда протекает именно так [46, 47]. Необходимо помнить, что повышенное АД часто проявляется болями в области сердца (по данным В. А. Азизова [35], частота синдрома стенокардии у больных АГ в сочетании с ИБС — 62,2 %, у больных АГ без ИБС — 41,7 %); при этом сахарный диабет будет способствовать возникновению субъективно неманифестированной ГБ.
Схема 1. Патогенез диабетической нейропатии.
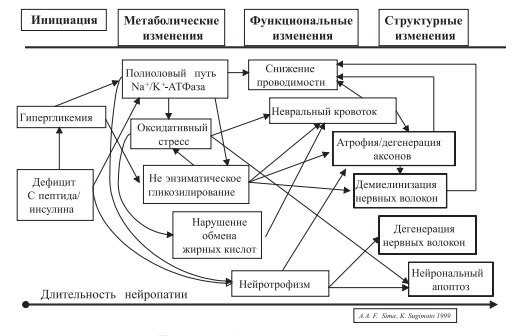
Потеря болевой чувствительности означает отсутствие лимитирующего фактора при нагрузке и, соответственно, повышает риск развития инфаркта миокарда и гипертонических кризов [47].
Многие исследования показали наличие очевидной связи между автономной нейропатией и гипертензией, что, вероятно, обусловлено значительной ролью в патогенезе гипертензии повышенной активности симпатического отдела нервной системы. Данные суточного мониторирования АД подтвердили наличие АГ у значительной части больных с автономной нейропатией, а степень тяжести последней коррелирует с уровнем повышения систолического и диастолического АД. Для диабетической автономной нейропатии (ДАН) характерно лабильное течение АГ с большим размахом колебаний АД в течение суток и снижением коэффициента средних значений АД день/ночь [48]. У больных с диабетом и ДАН наблюдается инверсия физиологического ритма АД, когда его ночные значения превышают дневные [50]. При ДАН развивается такой феномен, как артериальная гипертензия в положении лежа. Как правило, в этих случаях при длительном лежании днем или во время ночного сна отмечаются подъемы АДдо высоких цифр (180–220/100–120ммрт. ст.), которые, как правило, протекают бессимптомно и остаются незамеченными [51]. Эти сдвиги АД обусловлены так называемой постденервационной гиперчувствительностью адренорецепторов гладких мышц сосудов к медиаторам вегетативной нервной системы, которая формируется при хронических денервационных процессах (закон постденервационной гиперчувстви- тельности Canon). Механизм постденервационной гиперчувствительности до конца неясен. Ее можно рассматривать как результат адаптации чувствительности вегетативных эффекторов в условиях повреждения иннервирующих их постганглионарных волокон. Учет возможности появления артериальной гипертензии у больных, страдающих ортостатической гипотензией, является важным при назначении препаратов, повышающих АД.
Таким образом, при ассоциации гипертонической болезни с СД наблюдается целый ряд физиологических феноменов, определяющих повышение порога болевой чувствительности и создающих условия для десенситизации, что способствует отсутствию субъективной манифестации факта повышенного АД.
Особенности пожилого возраста, приводящие к развитию субъективно неманифестированной формы ГБ
Старение сердечно-сосудистой системы (ССС), так же, как и любой другой системы организма, является постоянным и необратимым процессом. В процессе старения наибольшее практическое значение имеют две основные группы изменений. К первой относятся прогрессирующие изменения морфологических, нейрогуморальных и функциональных характеристик ССС, ко второй — увеличение числа зависимых от возраста сердечно- сосудистых заболеваний, часто протекающих с атипичными проявлениями и сопровождающихся полиорганной недостаточностью и неблагоприятным прогнозом.
Клинические наблюдения в большинстве случаев свидетельствуют о том, что интенсивность болевого восприятия снижается с возрастом. Например, число случаев безболевых инфарктов увеличивается у пациентов старше 65 лет. Однако эти феномены могут объясняться различными особенностями проявления патологических процессов в пожилом возрасте, а не снижением болевого восприятия как такового. Тем не менее исследования величины порогов болевого восприятия у пожилых пациентов дают противоречивые результаты. По данным одних авторов, болевые пороги у пожилых пациентов повышаются [52], по данным других — понижаются [53] или остаются неизменными [54].
Так, Procacci [55] и другие исследователи [56] при определении у людей порогов болевой чувствительности с помощью теплового алгезиметра выявили закономерное снижение болевой чувствительности с возрастом. Результаты других работ свидетельствуют о достоверном повышении порогов болевой чувствительности у людей разных возрастных групп начиная с 30 лет. Т. М. Радзюкевич [57], Hakins и соавт. [58], изучая влияние возраста на восприятие боли при электростимуляции пульпы зуба у женщин различного возраста (средний возраст 22,2 и 79,3 года), не обнаружили повышения с возрастом болевых порогов. При моделировании патологической боли аппликацией капсаицина на кожу у молодых и престарелых людей возникала боль и гипералгезия одинаковой интенсивности. Однако у престарелых отмечался удлиненный латентный период до начала болевых ощущений и до развития максимальной интенсивности боли. У престарелых людей ощущение боли и гипералгезия длятся дольше, чем у молодых. Был сделан вывод, что у престарелых пациентов снижена пластичность ЦНС при длительном болевом раздражении. В клинических условиях это проявляется более медленным восстановлением и длительной повышенной болевой чувствительностью после повреждения тканей [58, 59].
Немаловажное значение имеет факт множественной патологической имульсации с периферических сенсоров в ЦНС, связанный с множественной ассоциированной возрастной патологией (обычно опорно-двигательного аппарата, сердечно-сосудстой системы, мочевыделительной системы), что, безусловно, снижает чувствительность пожилых пациентов к физиологическим раздражителям, таким как повышение АД, нагрузка на миокард и пр. В условиях поливалентной патологической импульсации, чаще всего болевой, пациенты вынуждены принимать аналгетические препараты, существенно понижающие болевое восприятие. Этому аспекту посвящена следующая часть статьи.
Патогенетические механизмы развития малосимптомных форм ГБ при длительном приеме анальгетиков
Механизм развития феномена снижения болевой чувствительности нельзя считать окончательно установленным. К развитию малосимптомных и бессимптомных форм ГБ может приводить ежедневное применение анальгетических средств (аспирин, ацетамифен, кодеин и др.), нестероидных противовоспалительных средств (амидопирин, анальгин, фенацетин, парацетамол, препараты салициловой кислоты, натрия салицилат, бутадион, ибупрофен, индометацин и пр.), эрготамина, дигидроэрготамина, суматриптана, барбитуратов, седативных препаратов и т. п. Наиболее активными в плане повышения порога болевой чувствительности считаются аспирин, кодеинсодержащие препараты, ацетамифен, кофеин, эрготамин. Имеет значение систематичность, длительность приема и дозы, превышающие обычные. Считают, что в результате избыточного употребления анальгетиков происходит угнетение центральной ноцицептивной системы [62]. Длительный прием анальгетиков угнетает образование и секрецию эндогенных аналгезирующих субстанций и ведет к снижению порога болевой рецепторной чувствительности рецепторов. Также угнетаются центральные структуры антиноцицептивной системы. В нервной системе имеются оn- и off-клетки, которые осуществляют контроль над болевой передачей. Оn-клетки активизируют ноцицепцию, off-клетки ингибируют ее. Установлено, что прием опиатных препаратов приводит к угнетению оn-клеток и активации off-клеток, что вызывает ослабление ноцицепции, которая сохраняется при отмене препаратов. Еще одной точкой приложения как возможная причина развития бессимптомных форм гипертонической болезни является пуриновая анальгезия. Реализация анальгетической активности пуринов опосредована рецепторами, называемыми пуриновыми или аденозиновыми, расположенными на постилсинаптических мембранах нейрональных или эффекторных клеток. В опубликованных эксперементальных исследованиях [64–66] продемонстрировано, что аденозин ингибирует залповую импульсацию как ноцицептивных, так и неноцицептивных нейронов задних рогов, которая считается отражением меры активации афферентных путей. Это ингибирование развивается, по крайней мере частично, за счет открытия калиевых каналов с последующей гиперполяризацией постсинаптической мембраны и формированием ингибиторного постсинаптического потенциала [66, 67]. Установленным фактом является противовоспалительное действие пуринов, которое развивается большей частью на периферии и вносит весомый вклад в их анальгетический эффект в целом. В результате срыва воспалительной вазоконстрикции вымываются медиа- торы воспаления — простагландины, гистамин, брадикинин, серотонин, субстанция Р и другие и невоспалительные альгогены — протоны, ионы калия, вызывающие феномен периферической сенситизации, что ведет к повышению порога возбуждения свободных нервных окончаний и деактивации молчащих ноцицепторов [65, 66].
Психологические особенности личности, приводящие к развитию субъективно-неманифестированной формы ГБ
Социальный и культурный уровень пациента, его психологические особенности оказывают существенное влияние на его способность к восприятию и качественной оценке потока ноцицептивной импульсации [33, 68].
Феномен отрицания является психологической защитой от угрожающей или крайне тревожной для пациента ситуации, позволяющей значительно уменьшить чувство страха и легче адаптироваться в социальном, физическом и психологическом отношении к качественно новой жизненной ситуации, сложившейся вследствие возникновения какого-либо заболевания или его осложнений. В целом, положительно влияя на процесс выздоровления, отрицание в ряде случаев приводит к недооценке пациентом состояния своего здоровья, игнорированию болезни [69]. Возможно, что у некоторых больных ГБ отрицание может обусловить появление бессимптомных форм.
Анализ пациентом причины возникших ощущений и их значимости для его здо- ровья оказывает значительное влияние на выраженность симптомов того или иного 31 заболевания. Как правило, люди избирательно прислушиваются к разнообразным сиг- налам, поступающим в ЦНС с поверхности тела или из внутренних органов, выделяя те, которые, по их мнению, основанному на предыдущем опыте или убеждениях, несут информацию о вероятном неблагополучии. Двусмысленные, неотчетливые ощущения, не совпадающие с их устоявшимися взглядами на причинно-следственные взаимосвязи в организме и состояние собственного здоровья, обычно игнорируются [70]. Так, больные ГБ со слабо выраженными клиническими проявлениями, ранее никогда не переносившие аналогичных ощущений, редко обращаются за медицинской помощью, ошибочно полагая, что появившиеся незначительные неприятные ощущения связаны с какими-то безвред- ными, преходящими изменениями в организме.
Полагают, что сниженная чувствительность к боли у человека может быть как следствием особых условий воспитания, так и наследственно детерминированной [15]. Клиницисты описывают лиц, у которых полностью отсутствует чувствительность к боли. Чаще всего это обусловлено врожденными дефектами развития периферических или центральных сенсорных путей — отсутствием рецепторов боли, определенных групп афферентных волокон в периферических нервах или спинномозговых ганглиях либо первого нейрона высшего порядка в заднем роге спинного мозга в тракте Лиссауэра [71].
Лица с такими дефектами слабо различают тепло — холод, острое — тупое, у них ослаблено потоотделение и слезотечение, отсутствуют роговичные и сухожильные рефлексы. Существуют также люди с врожденным безразличием к боли, у которых сенсорные пути и центральные нервные структуры развиты нормально, полностью сохранены другие сенсорные модальности, а также потоотделение, слезотечение, роговичные рефлексы. Такие лица воспринимают и правильно оценивают болевые воздействия, но не способны определить их опасность, а также реагировать на болевые стимулы защитным или агрес- сивным поведением. Полагают, что причина безразличия к боли лежит в поражении центральных интеграционных структур болевой системы [15].
В настоящее время в литературе активно обсуждается вклад гипотироза в течение артериальной гипертензии. По данным ряда исследований [72], гипотироз существенно влияет на повышенные уровни артериального давления, вызывает гипоалгезию, в частности, влияет на экспрессию периферических бензодиазепиновых рецепторов (PBRs), которые связаны с восприятием болевой чувствительности.
Таким образом, представленные в обзоре данные литературы показывают факт существования субъективно неманифестированного варианта течения гипертонической болезни, а также описывают наиболее вероятные механизмы развития феномена болевой десенситизации, ведущими из которых являются: гипертензивная гипоалгезия, ассоции- рованная сердечно-сосудистая и эндокринная патология, в частности — гипотироз, прием препаратов, влияющих на нервную передачу, пожилой возраст, психологические особен- ности личности, возможная пуринэргическая анальгезия.
Литература:
1. Оганов Р. Г. Методические вопросы профилактики сердечно-сосудистых заболеваний. М., 2002. 205 с.
2. Шальнова С. А., Баланова Ю. А., Константинов В. В. и др. Артериальная гипертония: распростра- ненность, осведомленность, прием антигипертензивных препаратов и эффективность лечения среди населения Российской Федерации // Рос. кардиол. журн. 2006. № 4. С. 45–50.
3. Кушаковский М. С. Эссенциальная гипертензия. СПб., 2002. 416 с.
4. Гогин Е. Е. Артериальные гипертонии: патогенетические механизмы и клиническая практика // Кар- диоваск. тер. и профил. 2003. № 4. С. 5–7.
5. Гогин Е. Е. Гипертоническая болезнь: основы патогенеза, диагностика и выбор лечения // Con. med. 2004. № 6. С. 324–330.
6. Moser M. A. Decade of progress in the management of hypertension // Hypertension. 1983. Vol. 5. P. 808–813.
7. Алексеева Л., Докина Е., Полубоярова Н., Кукушкин А. Трудности диагностики и лечения начальной стадии гипертонической болезни в поликлинических условиях // Врач. 2007. № 4. C. 6–9.
8. Гогин Е. Е., Гогин Г. Е. Гипертоническая болезнь и ассоциированные болезни системы кровообра- щения: основы патогенеза, диагностика и выбор лечения. М., 2006. 254 c.
9. Taylor B. K., Roderick R. E., Lezin E. St., Basbaum A. I. Hypoalgesia and hyperalgesia with inherited hypertension in the rat // Amer. J. Physiol. Regul. Integr. Comp Physiol. 2001. Vol. 280. № 2. P. 345–354.
10. Zamir N., Shuber E. Altered pain perception in hypertensive humans // Brain. Res. 1980. Vol. 201. P. 471–474.
11. Meller S., Lewis S., Brody M., Gebhart G. Nociceptive afferent vagal input is enhanced after transection of aortic depressor nerve // Hypertension. 1990. Vol. 15. Р. 797–802.
12. Thurston C. L., Randich A. Acute increases in arterial blood pressure produced by occlusion of the abdominal aorta induces antinociception: peripheral and central substrates // Brain. Res. 1990. Vol. 519. Р. 12–22.
13. Morgan M. M., Fields H. L. Activity of nociceptive modulatory neurons in the rostral ventromedial medulla associated with volume expansion-induced antinociception // Pain. 1993. Vol. 52. Р. 1–9.
14. Hosobuchi Y., Adams J., Linchitz R. Pain relief by electrical stimulation of central gray matter in humans and its reversal by naloxone // Science. 1977. Vol. 197. P. 183–186.
15. Watkins L. R., Thurston C. L., Fleshne M. Phenylephrine-induced antinociception: investigations of potential neural and endocrine bases // Brain. Res. 1990. Vol. 528. Р. 273–284.
16. Лиманский Ю. П. Физиология боли. Киев, 1986. С. 93.
17. Верткин А. Л., Мартынов И. В., Гасилин В. С. Безболевая ишемия миокарда. М., 1995. 88 c
18. Кондратьев В. В., Бочкарева Е. В., Кокурина Е. В. Безболевая ишемия миокарда: Современное состояние проблемы и клинически значимые аспекты ее развития. Механизмы формирования безболевой ишемии миокарда // Кардиология. 1997. № 2. С. 90–95.
19. Лиманский Ю. П. Основные принципы функциональной организации ноцицептивных и анти- ноцицептивных систем мозга // Физиол. журн. 1989. № 2. C. 110–121.
20. Сапронов Н. С., Федотова Ю. О. Гормоны гипоталамо-гипофизарно-тиреоидной системы и мозг. СПб., 2002. 184 с
21. Лиманский Ю. П., Лиманская Л. И. Проблема боли в современной медицине // Журн. практ. врача. 2001. № 2. C. 37–39.
22. McCubbin J. A., Bruehl S. Do endogenous opioids mediate the relationship between blood pressure and pain sensitivity in normotensives? // Pain. 1994. Vol. 57. P. 63–67.
23. McCubbin J. A., Helfer S. G., Switzer III F. S. e. a. Opioid Analgesia in Persons at Riskfor // Hypertens. Psychosom. Med. J. 2006. Vol. 68. N 1. P. 116–120.
24. Elbert T., Rockstroh B., Lutzenberger W. e. a. Baroreceptor stimulation alters pain sensation depending on tonic blood pressure // Psychophysiology. 1988. Vol. 25. P. 25–29.
25. Dworkin B. R., Elbert T., Rau H. e. a. HM Central effects of baroreceptor activation in humans: Attenuation of skeletal reflexes and pain perception // Proc. Nat. Acad. Sci. USA. 1994. Vol. 91. P. 6329–6333.
26. Guasti L., Zanotta D., Mainardi L. T. e. a. Hypertension-related hypoalgesia, autonomic function and spontaneous baroreflex sensitivity // Autonomic neurosci.: basic and clinical. 2002. Vol. 99. N 2. P. 127–133.
27. Zamir N., Maixner W. The relationship between cardiovascu-25.lar and pain regulatory systems // Ann. N. Y. Acad. Sci. 1986. Vol. 467. P. 371–384.
28. Sheps D. S., Bragdon E. E., Gray T. F. D. e. a. Relation between systemic hypertension and pain perception // Amer. J. Cardiol. 1992. Vol. 67. P. 70.
29. Ghione S., Rosa C., Mezzasalma L., Panattoni E. Arterial hypertension is associated with hypoalgesia in humans // Hypertension. 1988. Vol. 12. P. 491–497.
30. Ghione S. Hypertension-associated hypalgesia: Evidence in experimental animals and humans, pathophysiological mechanisms, and potential clinical consequences // Ibid. 1996. Vol. 28. P. 494–504.
31. Koltyn K. F. Exercise, hypoalgesia and blood pressure // Sports Med. 2006.Vol. 36. № 3. P. 207–214.
32. Rosa C., Vignocchi G., Panattoni E. e. a. Relationship between increased blood pressure and hypoalgesia: additional evidence for the existence of an abnormality of pain perception in arterial hypertension in humans//J. Hum. Hypertens. 1994. Vol. 8. P. 119–126.
33. Morley G. K., Moaradian Ad., Levine A. S. Mechanism of pain in diabetic peripheralneuropathy: Effect of glucose on pain perception inhumans // Amer. J. Med. 1984. Vol. 77. P. 79–82.
34. Falcone C., Auguadro C., Sconocchia R., Angoli L. Susceptibility to pain in hypertensive and normotensive patients with coronary artery disease // Hypertension. 1997. Vol. 30. P. 1279–1283.
35. Азизов В. А., Савченко А. П., Атьков О. Ю. Особенности клинического течения ИБС у больных с артериальной гипертензией // Кардиология. 1993. Т. 33. № 6. С. 38–41.
36. Анисимов А. Ю. Теоретические и клинические аспекты концепции болевого синдрома. Казань, 2001. 320 с.
37. Ness Т., Gebhart G. Visceral pain: a review of experimental studies // Pain. 1990. Vol. 41. P. 167–234
38. Ammons W. S., Blair R. W., Foreman R. D. Vagal afferent in hibition of spinothalamic cell to sympathetic afferents and bradykinin in the monkey // Circulat. Res. 1983. Vol. 53. P. 603–612.
39. Космачев А. А., Фроленков Г. И. Снижение порогов болевого ощущения и защитного сгибательного рефлекса у больных ишемической болезнью сердца // Кардиология. 1989. Т. 29. № 6. С. 58–59.
40. Fleg J. L., Gerstenblith G., Zonderman A. B. e. a. Prevalence and prognostic signcance of exerciseinduced silent myocardial ischemia detected by thallium scintigraphy and electrocardiography in asymptomatic volunteers // Circulation. 1990. Vol. 81. P. 428–436.
41. Михайлов С. С. Клиническая анатомия сердца. М., 1987. 228 с.
42. Kennedy H. L., Sprague M. K., Homan S. M. e. a. Natural history of potentially lethal ventricular arrhythmia's in patients treated with long-term anti arrhythmic drug therapy // Amer. J. Cardiol. 1989. Vol. 64. P. 1289–1297.
43. Kennedy H. L., Seiler S. M., Sprague M. K. e. a. Relation of silent myocardial ischemia after coronary artery bypass grafting to angiographic completeness of revascularization and long term prognosis // Ibid. 1990. Vol. 65. P. 14–22.
44. Kennedy H. L., Brooks M. M., Barker A. H. e. a. Beta-blocker therapy in the cardiac arrhythmia suppression trial cast investigators // Ibid. 1994. Vol. 74. P. 674–680.
45. Laird-Meeter K., ten Katen H. J., Brower R. W. e. a. Angina pectoris, one to 10 years after aortocoronary bypass surgery // Eur. Heart. J. 1983. Vol. 4. P. 678–686.
46. Egstrup K. Asymptomatic myocardial ischemia as a predictor of cardiac events after coronary artery bypass grafting for stable angina pectoris // Amer. J. Cardiol. 1988. Vol. 61. P. 248–252.
47. Чазова И. Е., Мычка В. Б. Профилактика, диагностика и лечение метаболического синдрома: Пособие для практикующих врачей. М., 2005. 48 с.
48. Kempler P. Neuropathies // Pathomechanism, clinical presentation, diagnosis, therapy. 2002. P. 208.
49. Ametov A. S., Barinov A., Duck P. J. e. a. The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid // Diabet. Care. 2003. Vol. 26. P. 770–776.
50. Балаболкин М. И., Чернышова Т. Е., Трусов В. В., Гурьева И. В. Диабетическая нейропатия: пато- генез, диагностика, классификация, прогностическое значение, лечение: Учебно-методич. пособие. М., 2003.105 c.
51. Строков И. А., Аметов С. А., Козлова Н. А., Галеев И. В. Клиника диабетической невропатии // Рус. мед. журн.1998. Т. 6. № 12. C. 787–801.
52. Sima A. A. F., Zhang W., Sugimoto K e. a. C-peptide prevents and improves chronic The I diabetic polyneuropathy in the BB/Wor rat // Diabetologia. 2001. Vol. 44. P. 44.
53. Chakour M. C., Gibson S. J., Bradbeer M., Helme R. D. The effect of age on A delta and C-fibrethermal pain perception // Pain. 1996. Vol. 64. P. 143–152.
54. Collins R., McMahon S. Blood pressure, antihypertensive drug treatment and the risk of stroke and coronary heart disease // Br. Med. Bull. 1994. Vol. 50. P. 272–298.
55. Procacci P., Bozza G., Buzzelli G., Corte M. D. The cutaneous pricking pain threshold in old age // Gerontol. Clin. 1970. Vol. 12. P. 213–218.
56. Минц А. Я., Лысенюк В. П. Биология старения: Кожная чувствительность: Руководство по физио- логии. Л., 1982. C. 479–492.
57. Радзюкевич Т. М. О порогах болевой чувствительности у рабочих виброопасных профессий и у практически здоровых людей различного возраста // Гигиена труда и проф. заболеваний. 1974. № 4. С. 10–12.
58. Harkins S. W., Chapman C. R. The perception of induced dental pain in young and elderly women // J. Gerontol. 1977. Vol. 32. P. 428–435.
59. Крутько В. Н., Донцов В. И. Системные механизмы и модели старения. М., 2008. 336 с
60. Решетняк В. К., Кукушкин М. Л. Возрастные и половые различия восприятия боли // Клинич. геронт. 2003. Т. 9. № 6. C. 34–38.
61. Morin-Papunen L. C., Vauhkonen I., Koivunen R. M. e. a. Endocrine and metabolic effects of metformin versus ethinyl estradiol-cyproterone acetate in obese women with polycystic ovary syndrome: a randomized study // J. Clin. Endocrinol. Metab. 2000. Vol. 85. P. 31–61.
62. Guinsburg R., de Araujo Peres C., Branco de Almeida M. F. e. a. Differences in pain expression between male and female newborn infants // Pain. 2000. Vol. 85. P. 127–133.
63. Bradshaw R., Ganley R., Jones W., Dyer P. High levels of dothistromin toxin produced by the forest pathogen Dothistroma pini // Mycolog. Res. 2000. Vol. 104. P. 325–332.
64. Hayashida M., Fukunaga A., Fukuda K. e. a. The characteristics of intravenous adenosineinduced antinociception in a rabbit model of acute nociceptive pain: a comparative study with remifentanil // Anesth. Analg. 2006. Vol. 103. № 4. P. 1004–1010.
65. Eisenach J. C., Eisenach J. C. Adenosine reduces glutamate release in rat spinal synaptosomes//Anesthesiology. 2005. Vol. 103. P. 1060–1065.
66. Карелов Е., Захаров Д. А., Лебединский К. М., Семено Д. А. Новые технологии в анестезиологии: пуриновая анальгезия // Вестн. С.-Петерб. ун-та. Сер. 11. 2008. Прил. к вып. 1.
67. Dirks J., Moiniche S., Hilsted K. L., Dahl J. B. Mechanisms of postoperative pain: clinical indications for a contribution of central neuronal sensitization // Anesthesiology. 2002. Vol. 97. N 6. P. 1591–1596.
68. Fields H. Sources of variability in the sensation of pain // Pain. 1988. Vol. 33. P. 195–200.
69. Levenson J., Mishra A., Hamer R., Hastillo A. Denial and medical outcome in unstable angina // Psychosom. Med. 1989. Vol. 51. P. 27–35.
70. Pennebaker J. The psychology of physical symptoms. New York,1982.
71. Физиология человека: В 3-х т. / Под ред. P. Шмидта, Г. Тевса: Пер. с англ. М., 2005. Т. 1. 323 с
72. Guasti L., Marino F., Cosentino M. e. a. Pain Perception, Blood Pressure Levels, and Peripheral Benzodiazepine Receptors in Patients Followed for Differentiated Thyroid Carcinoma: a longitudinal Study in Hypothyroidism and During Hormone Treatment // Pain. 2007. Vol. 23. P. 518–523.
Источник: Обрезан А.Г., Шункевич Т.Н., Е. В. Юрченко Е.В., Рындин Р.А. Основные механизмы, приводящие к формированию субъективно неманифестированной гипертонической болезни // Журнал "Вестник Санкт-петербургского университета Сер.11 вып. №2, 2009" с. 21-34
