Амилоидоз — системный процесс, характеризующийся сложным нарушением белкового и углеводного обмена, и сопровождающийся экстрацеллюлярным отложением особого гликопротеида, различного происхождения. На сегодняшний день проблема амилоидоза остается все еще малоизученной в связи с малой популяционной распространенностью, трудностями диагностики и лечения, а также неблагоприятным прогнозом. Амилоидоз представляет собой термин для описания болезней, которые имеют общую особенность: внеклеточное депонирование патологических нерастворимых фибриллярных белков в органах и тканях. Амилоид дает окраску с конгорот, проявляющуюся красным окрашиванием при обычной световой микроскопии, яблочно-зеленым (изумрудной зелени) при наблюдении в поляризованном свете.
Впоследствии с появлением электронной микроскопии был описан фибриллярный характер амилоида и характерная бета-складчатая конфигурация, ответственная, по современным представлениям, за типичную окраску амилоида. Большим успехом обернулось открытие, что легкие цепи иммуноглобулинов являются составными частями амилоида. В ходе дальнейших исследований было определено, что амилоидные фибриллы состоят из различных видов белков при вторичном и семейном амилоидозе, это открыло путь к разработке терапии, направленной на источник продукции предшественников фибрилл [7].
Единичные данные свидетельствуют о возможности поражения от 0,1% до 6,6% населения при анализе аутопсий. На данном этапе до конца не установлен круг заболеваний при которых встречается ами
лоидное поражение органов в первую очередь называют туберкулез и ревматоидный артрит. Установлено также, что амилоидные отложения определяются в хронических гнойных очагах — остеомиелит, бронхоэктатической болезни и других хронических легочных нагноениях (несмотря на успехи целенаправленного лечения), сифилисе, а также лимфогранулематозе, опухолях паренхимы почки, легкого, неспецифическом язвенном колите, болезнях Крона и Уиппла, длительно текущем септическом эндокардите и других, более редких заболеваниях (например, при медуллярном раке щитовидной железы). Заболеваемость амилоидозом в различных странах напрямую зависит от популяционного распространения выше упомянутых заболеваний и особенно от процента распространенности наследственных форм патологии: семейная средиземноморская лихорадка, наблюдаемая у армян, евреев, арабов, — аутосомно наследуемое по рецессивному типу заболевание. Достаточно часто амилоидоз встречается в Испании, Португалии, Израиле, вероятнее всего это связано с распространением наследственных его форм, в Новой Гвинее — с первичным и локализованным амилоидозом. Низкую распространенность амилоидоза в Японии, Индии возможно объяснить особенностью питания в этих странах.
В настоящее время многие авторы все чаще акцентируют внимание врачей на развитие амилоидоза у пожилых лиц (особенно у лиц старше 70–80 лет), когда по данным вскрытий амилоидоз мозга обнаруживается у 63%, амилоидоз аорты — у 50%, сердца — у 37%, поджелудочной железы — у 30% больных. Описываются также новые формы амилоидоза, в частности амилоидоз как отдаленное осложнение гемодиализа (dialysis-assosiated amyloidosis, AAD), характеризующийся деформирующей артропатией, синдромом запястного канала, костными дефектами [9, 12].
Амилоид является сложным гликопротеидом, в котором фибриллярные и глобулярные белки тесно связаны с полисахаридами. Содержание многих аминокислот в сывороточных, тканевых белках, гиалине, коллагене и амилоиде разняться. На данный момент доказанным является наличие в амилоиде белков, близких по своим свойствам к α1-, β-, γ-глобулинам, а также альбумина, фибриногена, большого количества нейраминовой кислоты [9, 13]. Из полисахаридов в состав амилоида входят галактоза и глюкоза, в меньших количествах — галактозамины, глюкозамины, манноза и фруктоза. Высокую устойчивость амилоида к различным воздействиям вероятнее всего можно объяснить прочной связью между белковыми и полисахаридными фракциями. Изучение структуры амилоида дало возможность для понимания наиболее вероятных причин и механизмов формирования амилоида.
Специфическое окрашивание депозитов амилоида конго красным (рис.1), йодом и особенно метиловым фиолетовым и тиофлавином-Т связывают с фибриллярным строением амилоида.
Рис. 1. Амилоидные отложения двенадцатиперстной кишки — Конго красный. Описание: отложения амилоида в собственной пластинке слизистой 12-перстной кишки. Амилоид неизвестной этиологии. Амилоид представлен перивазальными гомогенными включениями после фиксации конго красным.

Инфракрасный и рентгенокристаллографический анализы показали, что фибрилла состоит из полипетидных цепей с кросс-бетаконформацией, это определяет двойное лучепреломление при окраске конго красным [13,14].
В состав амилоида также входит P-компонент — белок, идентичный по своему строению гликопротеиду плазмы SAP. Р-компонент составляет 10–15% от общей массы тканевого амилоида. SAP продуцируется гепатоцитами и является острофазовым белком, его повышение в сыворотке крови можно отметить при ревматоидном артрите, опухолях, заболеваниях печени. Отсутствие специфичности этого нормального сывороточного белка объясняется тем, что он связывается с амилоидными фибриллами. В 40–50-е амилоидоз классифицировали в зависимости от органной локализации, в 60–70е на основании вероятностного этиолгического фактора: идиопатический (первичный), наследственный (генетический), приобретенный (вторичный, реактивный), старческий и локальный.
В настоящее время амилоидоз классифицируют в соответствии с характером белков – предшественников плазмы, формирующих фибриллярные депозиты.
В зависимости от плазменного предшественника в настоящее время принято выделять различные типы амилоидоза (табл. 1) [14].
Таблица 1. Классификация амилоидоза.

Для практикующего врача рабочая классификация амилоидоза включает первичный, вторичный, наследственный и старческий, т.к. при обнаружении амилоидоза часто встает вопрос о выборе тактики лечения. Окончательной общепринятой классификации амилоидоза еще нет. Так как пока еще описываются новые формы амилоидоза, примером может служить семейный транстиретиновый амилоидоз (ATTR), одна из наиболее частых форм наследственного амилоидоза в США. В основе ATTR-амилоидоза лежат мутантные формы транстиретина (транспортный белок для тироксина). Различных мутаций (замещение единичных аминокислот), вызывающих только амилоидную полинейропатию, известно более 80. Не до конца определились классификационные группы, это особенно относится к амилоидозу, связанному с диализом, трактуемому некоторыми авторами как самостоятельную разновидность первичного амилоидоза, другими — как вариант вторичного амилоидоза [7].
Рассмотрим самые часто встречающиеся типы амилоидоза.
Системный АА-амилоидоз является вторичным и встречается при таких заболеваниях, как ревматоидный артрит, туберкулез, а также при наследственных заболеваниях: периодической болезни, нефропатическом семейном амилоидозе с глухотой, лихорадкой, крапивнице. Описаны и идиопатические варианты. При АА-амилоидозе белок фибрилл амилоида образуется макрофагами (амилоидобластами) из гуморального предшественника — SAA. При резком возрастании синтеза SAA и увеличении концентрации его в крови, происходит усиленная внутриклеточная деградация SAA, из фрагментов которого на цитомембране собираются фибриллы амилоида. На конечной стадии развития амилоидоза фибриллы продуцируются во внеклеточном матриксе. Превращение белков-предшественников в амилоидные фибриллы — многофакторный процесс, различающийся при образовании амилоида тех или иных типов. Предполагается существование определенного пускового механизма, связанного со старением. Доказательством этому служит старческий амилоидоз сердца, при котором отмечается депонирование фибрилл, производных нормального транстиретина, встречающийся исключительно у пожилых людей. Дальнейшее изучение фибриллогенеза может помочь объяснить агрессивность болезни при некоторых амилоидогенных белках-предшественниках и медленную прогрессию при других. AL, ATTR и AA (вторичный амилоидоз) отличаются полностью по своему патогенезу. Однако у каждого типа определяются похожие клинические черты, которые могут предполагать одну форму болезни или другую.
Первичный или идиопатический (AL) амилоидоз. При AL типе основным белковым компонентом амилоидных фибрилл являются легкие λ(VI)– и κ(I) — цепи иммуноглобулинов. В эту групп входят идиопатичский амилоидоз; миелома и другие В-гемобластозы; локальный амилоидоз разных органов. По мнению многих авторов в основе АL-амилоидоза лежит плазмоклеточная дискразия, характеризующаяся усилением синтеза легких цепей иммуноглобулинов плазматическими или миеломными клетками. Синтез фибриллярного белка амилоида из легких цепей иммуноглобулинов осуществляют макрофаги, на поверхности которых происходит сборка амилоидных фибрилл. Болезнь, которая встречается при гемобластозных диспротеинемиях, приводящая к выпадению в тканях различных органов легких цепей иммуноглобулинов. В моче или из тканевых отложений пациентов с первичным амилоидозом было выделено более полусотни различных видов моноклональных белков. Генетические основы AL-амилоидоза в настоящее время исследуются. Использование молекулярной генетики дало возможность исследователям обнаружить моноклональные клетки, содержащие идентичную иммуноглобулиновому гену клональную перегруппировку в периферической крови пациента. AL-амилоидоз представляет собой генерализованный диспротеинемический процесс с преобладающим поражением того или иного органа. Наиболее часто поражаемые органы — почка и сердце. [8] Также встречается амилоидоз сопровождающийся инфильтрацией желудочно-кишечного тракта, что нередко может проявляться мальбсорбцией или псевдообструкцией. Эти признаки далее сочетаются с потерей в весе, связанной с системным процессом и отеком, а также сопутствующей почечной или сердечной недостаточностью. Часто встречается гепатомегалия. Селезеночная дисфункция , является достаточно частым признаком, проявляющемся в 24 % случаев. Другие органы обычно вовлекаются реже. Сосудистая инфильтрация ведет к геморрагическим осложнениям, которые проявляются типичными «енотовыми глазами» — признаком спонтанной периорбитальной пурпуры (рис. 2), вызываемой минимальной травмой типа чихания или трением глаз [13] .
Рис. 2. Периорбитальная пурпура.
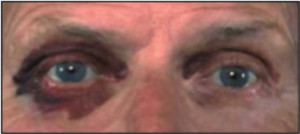
Массивные геморрагии при амилоидозе редкость и, когда оно происходит, нужно оценить факторы свертывающей системы крови. Макроглоссия выявляется приблизительно у 20% пациентов, характеризуется увеличением и уплотнением языка (рис. 3) [13].
Рис. 3. Увеличенный язык при амилоидозе.
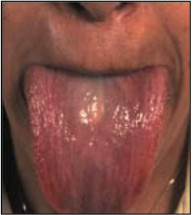
Язык часто обрамлен отпечатками зубов. Поднижнечелюстная припухлость типична вместе с увеличением языка и может быть достаточно большой, чтобы быть дыхательной преградой и вызывать феномен сонного апноэ. Инфильтрация мягких тканей амилоидом может происходить в любом месте, выражаясь в «плечевых подушках», дистрофии ногтей, или, редко, в алопеции. Также отмечаются расстройства вкуса иногда при отсутствии макроглоссии. Амилоидная инфильтрация голосовых связок приводит к охриплости или ослаблению голоса. В практике встречается и легочный амилоидоз, редко вызывающий клинические проявления. Инфильтрация надпочечников опосредует их гипофункцию, не всегда проявляющуюся клинически из-за других осложнений амилоидоза, таких как сердечная недостаточность, гипотензия и гипонатриемия вследствие дисфункции вегетативной нервной системы. Щитовидная железа может также быть инфильтрирована, и 10– 20 % пациентов имеют гипотиреоз.
ATTR-Амилоидоз, семейный (наследственный) амилоидоз характеризуется предрасположенностью этнических групп к этому заболеванию, особой географической распространенностью форм этого амилоидоза и наличием у родственников одной семьи. Транстиретин, транспортный белок для тироксина — белок с четырьмя идентичными субъединицами. Более пятидесяти различных замен одиночных аминокислот в транстиретине вызывают семейную амилоидную полиневропатию.
Семейные (генетические, наследственные) формы амилоидоза интенсивно изучаются в настоящее время. Среди этих форм описывают так называемый португальский амилоидоз, встречающийся у лиц португальского происхождения. Для этого типа свойственен аутосомно-доминантный тип наследования. У больных этим типом амилоидоза отмечают периферические нейропатии, нарушения функции кишечника, нарушения внутрисердечной проводимости, импотенцией. По данным ученых клиники Мейо амилоидные отложения в периферических нервных стволах могут вызывать нарушения функции желудочнокишечного тракта — дисфагия, чередование обстипации и диареи.[11,12] Описывают генетический нейропатический амилоидоз с поражением верхних конечностей — поражения кистей, карпорадиальный синдром.[10] Также скандинавские ученые описывают в четырех поколениях членов исландской семьи, 19 из которых умерли от геморрагического инсульта, при аутопсии амилоидные отложения были обнаружены в стенках мозговых артерий.
Амилоидоз при периодической болезни, передающейся аутосомно-рецессивно через несколько поколений семей, относится к АА-амилоидозу из-за антигенного сходства с сывороточным белком-предшественником SAA. Описываются и другие формы семейного амилоидоза: кардиопатический амилоидоз со смертью от прогрессирующей сердечной недостаточности (датский тип) [8]; нефропатический семейный амилоидоз с глухотой, лихорадкой, крапивницей и несколько других форм. АА (вторичный вариант амилоидоза) наиболее распространен и наиболее известен врачам. В отличие от первичного, наследственного, и старческого амилоидоза он развивается на фоне какого-либо заболевания. Клинические наблюдения, а также литературные данные свидетельствуют о том, что таким основным заболеванием может быть любое хроническое нагноение, инфекционное, инфекционно-аллергическое или опухолевое заболевание. В настоящее время наиболее частыми причинами развития вторичного амилоидоза являются ревматоидный артрит, хронические заболевания легких, туберкулез, остеомиелит, опухоли. Клиническая картина вторичного амилоидоза богата симптомами, что связано как с возможностью многообразных проявлений тех заболеваний, при которых развивается амилоидоз, а также особенностями поражения внутренних органов (прежде всего почек) амилоидом.
AS-амилоидоз (S-senile, старческий) — предшественником данного типа амилоидного белка является преальбумин. Впервые этот тип амилоидного белка был выделен Вестмарком в 1978 г. Старческий амилоидоз имеет широкое популяционное распространение, у лиц старше 60–80 лет составляет 30% и почти у 80% лиц старше 80–90 лет. Эта форма амилоидоза чаще выявляется у женщин. При этой форме отмечают поражения сердца, поджелудочной железы и сосудов головного мозга, что обуславливает во многих случаях старческую психическую и физическую деградацию, хотя нередко старческий амилоидоз протекает субклинически. Амилоидоз почек и других органов при этой форме встречается редко.
В 80-х годах было описано частое выявление синдрома запястного канала, причиной которого было отложение амилоида в синовиальной оболочке, сухожилиях у людей находящихся длительно на гемодиализе. Данная специфическая форма получила название амилоидоза, ассоциированного с диализом (dialis-associated amyloidosis). Предшественником этого типа амилоидного белка является β-2-микроглобулин и его фрагменты. Частота выявления диализного амилоидоза и выраженность его клинических проявлений зависят от длительности диализной терапии. Первые клинически проявления отмечаются а пациентов с 5 летним стажем, а 90% страдает через 15 лет гемодиализа. Самым частым клиническим проявлением является синдром запястного канала. Клиническая симптоматика проявляется болями, онемением, парестезией большого, указательного и среднего пальцев, связанная с отложением амилоидных масс в связочном аппарате кистей и развитием тендовагинитов сгибателей пальцев рук. При амилоидозе ассоциированным с гемодиализом могут наблюдаться синдром деструктивной артропатии (плечелопаточный периартериит, деструктивная спондилоартропатия), а также синдром остеопатии (кистозные поражения костной ткани, патологические переломы костей) (рис. 4) [9].
Рис. 4. Рентгенограмма костных кист или карпорадиальный синдром.
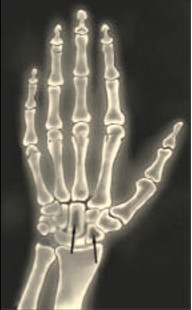
Диагностика амилоидоза основана на возможности клинически заподозрить наличие амилоидного поражения того или иного органа и установить его с помощью биопсии ткани. Когда клинически вероятность амилоидоза высока, исходя из комплекса клинических данных и возможной генетической предрасположености, то самая простая процедура — это получение образца подкожной жировой клетчатки живота. Аспират жира, окрашенный конго красным, будет позитивен с достоверностью в 85% у пациентов с AL-амилоидозом, хотя требуется опыт, чтобы избежать избыточной окраски ткани [13,14].
При отсутствии лечения прогноз амилоидоза неблагоприятен. И зависит от степени и количества пораженных органов. В среднем пациенты с AL амилоидозом имеют самый плохой прогноз, со средней выживаемостью от одного до двух лет. Пациенты с ATTR амилоидозом могут жить до 15 лет, а прогноз для пациентов с AA амилоидозом часто определяется во многом основным хроническим заболеванием.
Лечение амилоидоза направлено и на пораженный орган, и на специфический тип заболевания. Включает в себя коррекцию различных звеньев заболевания: устранение факторов, способствующих образованию амилоида, торможение продукции амилоида, воздействие на амилоидные отложения и симптоматическую терапию.
Симптоматическая терапия зависит от характера и степени нарушения пораженного органа, а предотвращение отложения амилоида от вида первичного заболевания. Однако существую препараты которые рекомендуются для приема при различных формах амилоидоза. При семейной Средиземноморской лихорадке, генетическом нарушении, сочетающемся с высокой частотой возникновения AA амилоидоза, терапия колхицином специфично воздействует на вызывающую болезнь и предотвращает амилоидоз. Предполагают, что колхицин ингибирует синтез SAA-протеина гепатоцитами. В связи с этим колхицин широко используют всех формах амилоидоза начиная с небольших доз, постепенно доводя дозировку до 2 мг/сут. в зависимости от индивидуальной переносимости. Существуют клинические исследования в которых использовали различные режимы прерывистого перорального приема мелфалана и преднизолона, и была доказана эффективность этой терапии в сравнении с группой не получавшей лечения, а также при терапии одним колхицином. В литературе также описываются схемы внутривенного введения мелфалана, но учитывая высокую цитотоксичность препарата широта использования внутривенной терапии остается под вопросом. При химиотерапии иодинатом антрациклина пять из восьми пациентов имели очевидное клиническое улучшение, включая уменьшение инфильтрации кожи и диареи. Учитывая, что транстиретин преобладающе синтезируется в печени, в качестве терапии для этого заболевания используют трансплантацию печени. Трансплантация печени ведет к исчезновению мутантного транстиретина из крови и улучшение течения невропатии. У диализных больных прибегают к хирургическому удалению тканей с амилоидными отложениями и симптоматической терапии. [13,14].
Теперь хотелось бы представить случай из практики классического AL амилоидоза развившегося на фоне миеломной болезни. Наблюдение нами пациентки почти в течение 10 лет позволило проследить классическую картину амилоидоза, его прогноз и исход. Пациентка в течение нескольких лет страдала миеломной болезнью и кроме этого множественной коморбидной патологией (сердечно-сосудистой, цереброваскулярной, эндокринными нарушениями и костномышечной патологией). Миеломная болезнь была диагностирована после перелома шейки бедра при травматическом факторе незначительной выраженности. Впоследствии прогрессирующая сердечная недостаточность, прогрессирование центральных и периферических неврологических нарушений, дисфагия, запоры, чередующиеся с диареей, возраст пациентки, а также наличие миеломной болезни позволили заподозрить амилоидное поражение центральной и периферической системы, сердца, почек, желудочно-кишечного тракта.
Пациентка Р., 88 лет, на момент рассмотрения по вышеуказанному поводу более 10 лет страдала ишемической болезнью сердца, атеросклерозом коронарных артерий, аорты, атеросклеротическим кардиосклерозом, осложненным прогрессирующей ХСН. У нее периодически возникали нарушения ритма сердца по типу наджелудочковой и желудочковой экстрасистолии, пароксизмов наджелудочковой тахикардии, пароксизмов фибрилляций предсердий. В течение последних лет принимала следующие препараты: бетаадрено-блокаторы, ингибиторы ангиотензинпревращающего фермента, антиагреганты, статины. Также были диагностированы многочисленные сопутствующие заболевания. Более 15 лет отмечала периодические повышения АД до 180\100, при резкой перемене погоды, рабочим давлением считала уровни 120\80. Верифицированы: атеросклероз сонных артерий (множественные стенозы ОСА, ВСА, НСА), цереброваскулярная болезнь, атеросклероз сосудов головного мозга, хроническая недостаточность мозгового кровообращения в вертебрально-базилярном бассейне. С 2004 года установлена дисциркуляторная энцефалопатия 3 ст., декомпенсация. В течение нескольких лет до описываемых событий наблюдалась также по поводу многоузлового зоба 2 степени, состояние эутиреоза. Из костно-суставной патологии отмечены остеоартроз правого коленного сустава II-III степени, дегенеративное изменение мениска правого коленного сустава.
Одним из значимых медицинских событий в жизни пациентки с приведенным комплексом патологии явился случайный перелом шейки правой бедренной кости. По этому факту 26.02.2006 г выполнена операция тотального эндопротезирования правого тазобедренного сустава (рис. 5).
Рис. 5. Рентгенограмма тазобедренного сустава после операции тотального эндопротезирования правого тазобедренного сустава от 26.02.2006г.
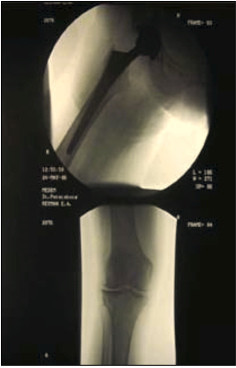
Среди вариантов сопутствующей патологии обращали на себя внимание варикозная болезнь; хроническая венозная недостаточность — I степени, диагностированные с 2003 года.
Также в 2005 году гематологом верифицирована анемия нормохромная, нормоцитарная, легкой степени, смешанного генеза: железодефицитная и В12-дефицитная.
Во время госпитализации в январе 2007 года, проводившейся по поводу нарушений сердечного ритма, в анализе крови обратило на себя внимание прогрессирование анемического синдрома (со сниженным количеством эритроцитов до 3,61*1012/л; с лейкопенией до 3,5*109/л; гемоглобином 112 г/л, относительным лимфоцитозом 58,8%, умеренной тромбоцитопенией — 162*109/л). Ретроспективное рассмотрение показателей клинического анализа крови выявило интересную динамику параметров эритроцитарного и лейкоцитарного рядов: постепенное снижение числа эритроцитов, лейкоцитов и скачкообразный рост количества лимфоцитов, значительное увеличение СОЭ (табл. 2).
Таблица 2. Ретроспективный анализ показателей клинического анализа крови пациентки.
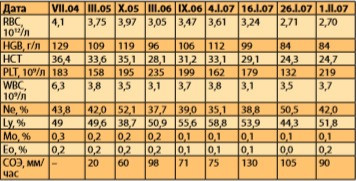
Учитывая анемический синдром, было проведено дополнительное обследование для исключения новообразования внутренних органов (анализ онкомаркеров, УЗИ внутренних органов). Также выявлены анамнестические сведения о консультации пациентки гематологом в апреле 2005 года: с учетом нормального содержания железа, фолиевой кислоты и витамина В12 в сыворотке крови, отсутствия данных за онкологическую патологию, положительного эффекта от проведенной общеукрепляющей терапии, нормальных показателей ферритина и железо-связывающей способности сыворотки крови было высказано предположение о возможном дефиците пластических элементов кроветворения в костном мозге и выставлен диагноз анемии смешанного генеза (В12 и железодефицитная) легкой степени; рекомендовано проведение терапии препаратами железа и витамина В12 в течение 1 месяца с целью восполнения костномозгового депо. Указанный вариант терапии не был эффективен. Позже проведен скрининг аутоиммунных заболеваний как вероятных причин анемического синдрома: какихлибо специфических антител к форменным элементам крови или их пластическим компонентам не было выявлено.
С учетом нарастающего анемического синдрома было выполнено УЗИ и КТ органов брюшной полости: данных за увеличение и изменение структуры паренхиматозных органов живота не выявлено. В заключении по лучевым методам исследования значилось: признаки хронического атрофического панкреатита, хронического холецистита, кисты правой почки; признаки мелких кистозных образований матки; КТ — признаки грыжи пищеводного отверстия диафрагмы.
При объективном исследовании определялись кожа и видимые слизистые бледные, чистые. Признаков геморрагического синдрома не выявлялось. Увеличенных периферических лимфатических узлов гепато– и спленомегалии не выявлялось. В гемограмме сохранялась трехростковая цитопения, нейтропения, относительный лимфоцитоз, определялась нормохромная, нормоцитарная анемия легкой степени. При анализе всех данных, лабораторно-инструментальных исследований было выявлено постепенное повышение общего белка (табл. 3).
Таблица 3. Динамика концентрации общего белка крови пациентки.

При дополнительном исследовании белков крови методом электрофореза выявлен М-градиент в гамма-спектре, альбумины были снижены до 32,6%, глобулины повышены до 67,4%, за счет гамма глобулинов 46,7% (таб. 4).
Таблица 4. Фракции белка крови пациентки.

На основании приведенных данных сделано предположение о наличии у пациентки дебюта одного из нижеуказанных заболеваний: парапротеинемического гемобластоза в варианте множественной миеломы, макроглобулинемии Вальденстрема, секретирующей лимфомы. Также проводился дифференциальный диагноз с группой моноклональных гаммапатий неутонченного первичного генеза.
С целью оценки костномозгового кроветворения под местной анестезией 1% раствором лидокаина была выполнена аспирационная биопсия костного мозга. Из задней верхней ости левой подвздошной кости получен 1 мл костномозгового пунктата для цитогенетического и цитологического исследования. В миелограмме был выявлен плазмоцитоз костного мозга до 72,6%.
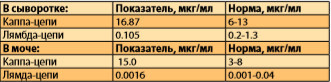
При дальнейшем обследовании: суточная потеря белка составляла 387,3 (при норме до 141), концентрации Ig А и Ig М были снижены до 0,22 г/л (при норме не ниже 0,25 г/л) и до 0,33 г/л (при норме не ниже 0,4 г/л) соответственно, а уровень Ig G составил 48,74 г/л (при норме 7-16 г/л). В исследовании легких цепей Ig было обнаружено увеличение числа каппа цепей в сыворотке крови (до 16,87 мкг/мл), и в моче (до 15,0 мкг/мл) (табл. 5) .
Таблица 5. Данные исследования легких цепей Ig.
На тот момент были также выполнены рентгенограммы плоских костей: черепа, костей таза, ребер. На рентгенограмме черепа вблизи венечного шва справа в лобной кости определялся округлый участок разрежения костной ткани с умеренно склерозированным контуром, более вероятно — пахионова грануляция, но нельзя исключить наличие солитарного миеломного очага (рис. 6).
Рис. 6. Рентгенограмма черепа, солитарный миеломный очаг (указан стрелкой).

C учетом плазмоцитоза костного мозга (72,6%), парапротеинемии, увеличения Ig G до 47 г/л, выявления легких цепей класса каппа, был выставлен окончательный диагноз: Множественная миелома, диффузно очаговая форма, III А стадия, тип G, класс каппа. Миеломная нефропатия ХПН-О.
Учитывая возраст пациентки, характер сопутствующей патологии, была рекомендована низкоинтенсивная стандартная химиотерапия первой линии по программе МП — мелфалан 9 мг/м2/сутки внутрь в течение 5 дней + преднизолон 0,5 мг/кг внутрь в течение 5 дней; каждые 4 недели. Определенный интерес вызывает предложенная сопроводительная терапия ассоциированных с проводимым лечением проявлений: аллопуринол 0,1–3 раза в сутки, гидратация 1л/м2/сутки (с целью ощелачивания мочи), зомета 4 мг в/в капельно раз в 4 недели, интраглобин 2 мл/кг 100-200 мл в/в капельно в течение 6-8 часов каждые 4 недели, гемокомпонентная терапия — переливание эритроцитарной массы при снижении Hb < 80 г/л. Первый курс лечения (и 10-15 дней после окончания химиотерапии) предложено провести в условиях стационара.
Ведущее значение в состоянии больной имело гематологическое заболевание — парапротеиноз, протекающий с синдромами анемии, нейтропении, иммунодефицита, гипервязкости, остеопении, и др. Первый курс терапии мелфаланом с 10 по 16 апреля 2007г., второй — с 08 по 14 июня 2007г. Третий курс с 24 по 28 июля 2007., четвертый курс с 10 по 15 сентября 2007 г. Из токсических эффектов наблюдалась постцитотоксическая панцитопения с развитием вторичного иммунодефицитного состояния. Проводилась колониестимулирующая терапия (граноцит — под контролем картины крови), на фоне которой временно уменьшалась степень нейтропении, анемии.
Учитывая выраженную индуцированную предыдущими курсами лейкопению, нейтропению, наличие сыпи на лице и геморрагий на ногах рассматривались как бактериальное и токсическое проявления вышеуказанных синдромов иммунодефицита, гипервязкозти, нейтропении. Пациентка в течение 2 недель наблюдалась в условиях стационара с содержанием в изолированной палате с повышенным эпидемиологическим режимом, дезобработкой палаты, УФО воздуха.
Повторное поступление было вызвано с закрытым перипротезным переломом верхней\средней трети диафиза правого бедра со смещением отломков. Проведена операция репротезирования.
В послеоперационном периоде обращали внимание прогрессирование диспепсических явлений и нарушений когнитивной функции, ухудшение фильтрационной функции почек. После недели пребывания в стационаре пациентка была переведена в ОРИТ в связи с общим ухудшением состояния, что было преимущественно связано с гиповолемией, нарушением ритма по типу фибрилляции предсердий (ФП) и анемией до Hb 60 г/л, подозрением на динамическую (обструктивную) кишечную непроходимость, частыми позывами на рвоту. Пациентке был установлен назогастральный зонд в связи с анорексией, тошнотой и рвотой при приеме жидкой пищи, кишечной непроходимостью, что в последствии трактовалось как проявления амилоидного поражения кишечника, периферической и центральной нервной системы. Грубой очаговой симптоматики не выявлялось, нарушений акта глотания не было. Отказ от приема связан с негативизмом пациентки на фоне выраженных когнитивных нарушений, вероятно обусловленных амилоидозом центральной нервной системы.
При проведении диагностической фиброколоноскопии были получены данные, свидетельствующие об амилоидном поражении прямой кишки, дивертикулезе долихосигмы, полипозе толстой кишки, комбинированном геморрое. Впоследствии гистологическое исследование участков толстой кишки, забранных при фиброколоноскопии, подтвердило наличие большого числа амилоидных отложений.
Исходя из вышеперечисленных данных клинической картины заболевания (синдром общемозговых дисфункций; дисфагический синдром; диарея чередующееся с обстипацией; нарастание признаков СН и ХПН) и лабораторно-инструментальных данных пациентки, можно достоверно судить об амилоидном поражении кишечника, и косвенно судить о поражении сердца, почек и нервной системы. В итоге можно проследить развитие классического AL-амилоидоза на фоне миеломной болезни, с поражением большого количества органов, и неблагоприятным прогнозом.
Литература:
1. Справочник гематолога. A-Z/ Бэйн Б. Дж., Гупта Р.; Пер. с англ. Мосоловой Т. П.; Под ред. Рукавицына О. А. — М.:БИНОМ. Лаборатория знаний, 2004. — 280 с., ил.
2. Руководство по гематологии: в 3 т. Т. 2. Под ред. Воробьева А. И. 3-е изд., перераб. и допол. М.: Ньюдиамед; 2003. — 280 с., с ил.
3. Абдулкадыров К. М. Клиническая гематология: Справочник. — СПб: Питер, 2006. — 448 с. — (Серия «Спутник врача»).
4. Окороков А. Н. Диагностика болезней внутренних органов: Т. 4. Диагностика болезней системы крови: — М.: Мед. лит., 2001. — 512 с.: ил.
5. Болезни системы крови (Файнштейн Ф. Э., Козинец Г. И., Бахоамов С. М., Хохлова М. П.; Прелисл. Гаврилова О. К.). — 2-е изд., перераб. и доп. — Т.: Медицина, 1987 — 671 с.: рис. табл.
6. Окороков А. Н. Лечение болезней внутренних органов: Т. 3, кн. 2. Лечение болезней сердца и сосудов. Лечение болезней системы крови: — М.: Мед. лит., 2001. — 480 с.: ил.
7. Системный первичный (идиопатический) амилоидоз. А. П. Ивкова, Г. Е. Гендлин, Г. И. Сторожаков, И. Г. Никитин, М. П. Пружковская \ Российский медицинский журнал, (2007), 2 (апрель). — С. 47-49.
8. Амилоидоз сердца. Светлана Ивановна Овчаренко, Сон, Окишева, Владимир Иванович Маколкин. — Клиницист, (2007), 6 , 32-38.
9. β2 -Microglobulin-selective Adsorbent Column (Lixelle) for the Treatment of Dialysis-related Amyloidosis \\ Kazuo Suzuki, Masami Shimazaki, Hidetoshi Kutsuki \\ Therapeutic Apheresis, 7 (2003), 1 , 104-107.
10. A peculiar form of peripheral neuropathy: familiar atypical generalized amyloidosis with special involvement of the peripheral nerves Andrade, Corino. \\ Brain; a journal of neurology, 75 (1952), 3 , 408.
11. Acute small bowel pseudo-obstruction due to AL amyloidosis: a case report and literature review R.N. Koppelman, N.H. Stollman, F. Baigorri, A.I. Rogers \\ The American Journal of Gastroenterology, 95 (2000), 1 (январь), 294-296.
12. S Tada, M Iida, T Yao, T Kitamoto, T Yao, M Fujishima Intestinal pseudo-obstruction in patients with amyloidosis: clinicopathologic differences between chemical types of amyloid protein. \\ BMJ Publishing Group Ltd & British Society of Gastroenterology 1993;34:1412-1417; doi:10.1136/gut.34.10.1412.
13. References: Mayo Clinic Proc. 2006;81(7):880-888.
Источник: Обрезан А.Г., Стрельников А.А., Косарев М.М. Особенности диагностики клинического течения амилоидоза случай из практики // Журнал медицина. XXI век № 4 {13} 2008, с. 63-70
